Welcome to dftfit’s documentation!¶
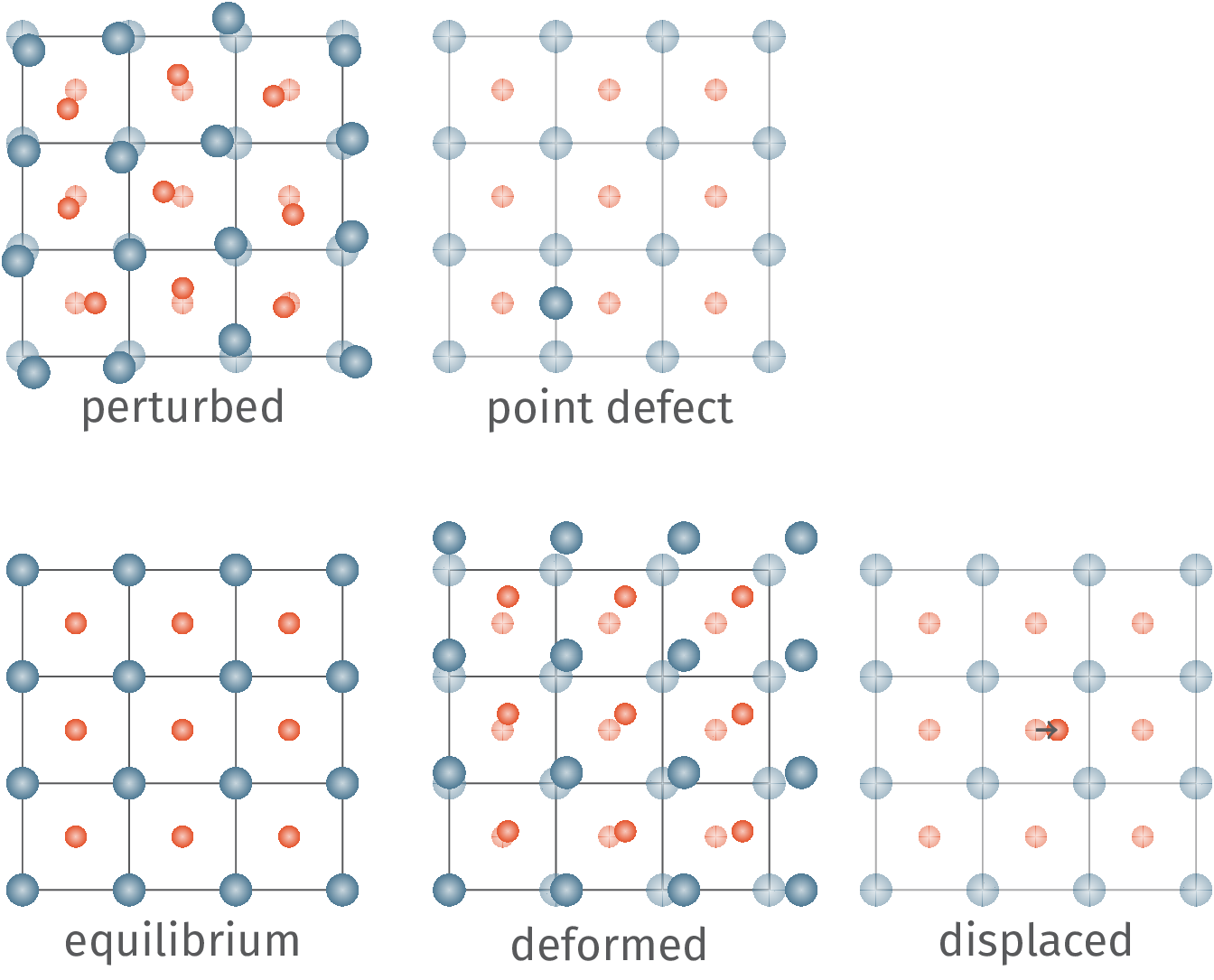
DFTFIT is a python code that used Ab Initio data from DFT calculations such as VASP, Quantum Espresso, and Siesta to develop molecular dynamic potentials. Our package differs from other similar codes in that we leverage LAMMPS as a calculator enabling a wide variety of potentials. The potentials include custom python functions and a wide variety or three-body interactions including the Tersoff, Stillinger-Weber, Gao-Weber, Vashishta, and COMB Potentials. All of which can be combined to have for example a Buckingham + Coulomb + ZBL potential. We also have an extensive set of multi-objective and single-objective optimizers that can evaluate a potential for many properties including energy, forces, stress, lattice constants, elastic constants, bulk modulus, and shear modulus.
- In general three things are required from the user.
- Ab-Initio Training Data includes VASP, Siesta, and Quantum Espresso Calculations. Additionally the user may supply measured properties such as lattice constants, elastic constants, bulk modulus, and shear modulus.
- configuration specifies optimization algorithm and number of steps, sqlite database to store results, MD calculator to use, weights to give for each property.
- Potential among a rich set of two and three body potentials. Including a custom python function.